
2021年7月30日
医療機器の薬事に関する法規制情報提供サービス

【法規制解説(日本)】医療機器を開発・販売するには?
ヘルスケアに関する技術は日に日に進歩しています。
特にソフトウェアの進歩はめざましいものがあります。
例えば、2020年にはApple Watchの心電・心拍測定機能アプリが医療機器承認を取得し、話題となりました。
それまでの心電計や心拍測定装置と言えば胸に電極パッドを当てて安静にして測定するものというイメージでしたが、いまや腕時計で測定できるようになったのですね。
Apple Watchは、そもそもはスマートウォッチと呼ばれる腕時計の一種です。
それが技術の進歩により医療機器としての機能を有するようになり、ついに「医療機器」として承認されるようになりました。
このように、技術革新に伴い医療機器産業に進出する方々や企業が増えてきています。
医療機器を市販するにあたって新規参入の方々にとっての障壁となるのは、医療機器に係る規制でしょう。 この記事では、日本で医療機器を作って市場で販売するためにクリアすべき規制要件について易しく解説します。
目次
簡単なフロー

Step1 開発しようとしている機器が医療機器に該当するか確認する
まずは開発しようとしている機器が医療機器に該当するかどうか判断します。
作ろうとしている機器が以下のどちらかに当てはまるのであれば、それは医療機器にあたる可能性があります
- 人または動物の病気の診断、治療もしくは予防に使用する機器
- 人または動物の身体の構造もしくは機能に影響を及ぼすことを目的とする機器
他社先行品がない等、医療機器に該当するかどうか分からない場合には、事業所を構えている都道府県の薬務課に相談してください。薬務課が、当該機器が医療機器に該当するかどうか、厚生労働省に確認してくれます。
また、プログラムが医療機器に該当するかどうかについては「プログラムの医療機器該当性に関するガイドライン」が発出されています。
上記ガイドラインを参照した上で、迷うようであれば事業所が所在する都道府県の薬務課にご相談ください。
Step2 開発しようとしている医療機器のクラスを確認する
次に作ろうとしている医療機器のクラスを確認してください。
医療機器は身体に与えるリスクによって4つのクラスに分類されます。
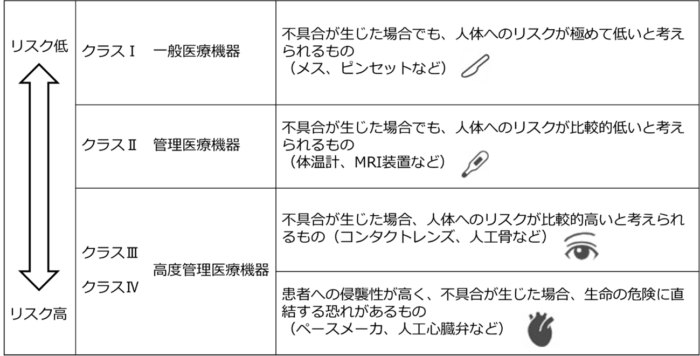
医療機器のクラスに応じて、その後対応すべき規制要求事項が変わってきます。
作ろうとしている医療機器の一般的名称(=国が定めた医療機器の名前)が分かっている場合には、PMDAデータベースからクラスや適用される規制要求事項を調べることができます。
Step3 QMS省令その他の準拠すべき基準に則って設計開発する
医療機器は開発過程から国の規制を受けます。
医療機器の開発はQMS省令(厚生労働省令第169号)を遵守して行う必要があります。
簡単に言うと、設計開発に関する手順書を作成し、その手順書に従って設計・開発を行います。
そして、そのとおりに実施したという証拠(=記録)を作成します。
その記録が医療機器の認証や承認のための審査を受ける際に、当局などから調査されます。
また、ISO14971に準拠したリスクマネジメントや、プログラムを含む医療機器の場合にはIEC62304に準拠した開発が求められます。
認証によって販売が認められる医療機器に関しては、医療機器ごとに準拠すべき基準(=認証基準)がありますので、PMDAデータベースなどでご確認ください。
販売のために国の承認を必要とする、基準の定められていない医療機器の場合には、どのようにして当該医療機器の安全性や有効性を示せばよいか、PMDAにご相談ください。
医療機器によっては治験が必要となる場合もあります。
Step4 製造販売業許可を取得する・製造業登録を行う
作ろうとしている医療機器を市場で販売するためには、関係する業者が製造販売業許可を取得したり、製造業登録を行う必要があります。
これ以降、「製造販売」という言葉を使用します。 製造販売とは、簡単に言うと「作って販売する」という意味です。
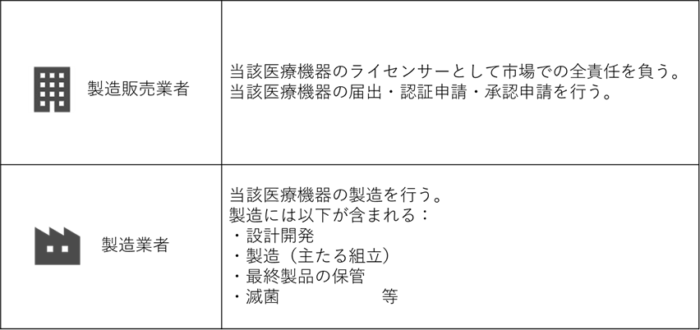
製造販売業許可について
製造販売業許可には3つの種類があります。
以下のように、Step2で特定した製造販売しようとする医療機器のクラスに応じて、適切な許可を取得する必要があります:
- 第1種医療機器製造販売業 高度管理医療機器の製造販売を行う業者
- 第2種医療機器製造販売業 管理医療機器の製造販売を行う業者
- 第3種医療機器製造販売業 一般医療機器の製造販売を行う業者
製造販売業許可の取得のためには、以下を整えることが必要です。
(1)人的要件
以下の5役を置く必要があります:
- 管理監督者
- 管理責任者
- 医療機器等総括製造販売責任者
- 国内品質業務運営責任者
- 安全管理責任者
上記のうち、「医療機器等総括製造販売責任者」「国内品質業務運営責任者」「安全管理責任者」にはそれぞれその役職に就くための資格要件が法令によって規定されています。
資格要件については以下の記事をご参照ください。
また、「5役」を置かなければならない=当該役職を担当する人数が「5人」必要、というわけではありません。
取得する製造販売業の種類に応じて、上記5役のうち複数の役職の兼務も可能です。
5役の兼務が可能な範囲については以下の記事をご参照ください。
(2)QMS体制省令への適合
「医療機器又は体外診断用医薬品の製造管理又は品質管理に係る業務を行う体制の基準に関する省令」(厚生労働省令第94号)に適合する必要があります。
簡単に言うと、以下が求められています:
- QMS省令を遵守したQMS体制を確立すること
- (1)人的要件に挙げた5役のうち①~④を設置すること
(3)GVP省令への適合
「医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準に関する省令」(厚生労働省令第135号)に適合した市販後安全管理体制を整える必要があります。
簡単に言うと、以下が求められています:
- GVP省令で求められる手順書を確立し、市販後安全管理を実施する体制を整える
- (1)人的要件に挙げた5役のうち、⑤安全管理責任者を設置する
- 第1種医療機器製造販売業の場合には安全管理統括部門を設置する
(1)~(3)を整えた上で、事業所の存する都道府県に製造販売業許可取得のための申請を行ってください。
都道府県の担当者が(1)~(3)が適切に整えられているかどうか実地調査を行い、適合が確認された場合には業許可が付与されます。
製造業登録について
製造販売しようとする医療機器について、以下を実施する業者は製造業者登録が必要になります:
- 設計開発
- 主たる組立
- 最終製品の保管
- 滅菌
製造業者登録をするためには、責任技術者を設置した上で、事業所の存する都道府県に登録手続きを行う必要があります。
許可制ではないので簡素な手続きのみで済みますが、都道府県によっては事業所の実地確認を行う場合があります。
また、登録制であるためQMS省令を遵守したQMS体制の確立はこの時点では求められていませんが、Step5の認証・承認申請を行った際の調査で製造業者のQMSも調査されます。
そのため、製造業者もQMS省令に則ったQMS体制を確立しておく必要があります。
Step5 設計開発した医療機器の届出・認証申請・承認申請を行う
いよいよ最後のステップです。
医療機器のクラスに応じた製造販売のための手続きを行います。
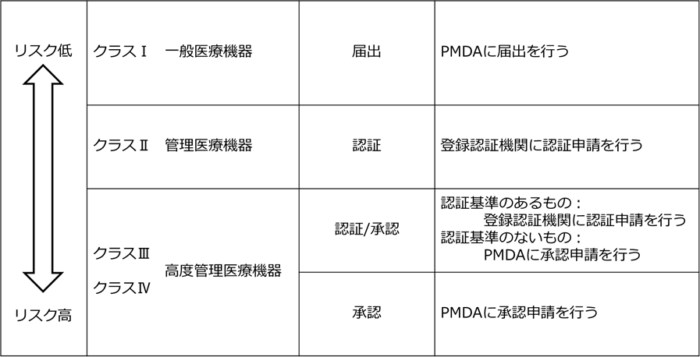
クラスⅠ医療機器の場合は、PMDAに届出を行い、受理されればすぐに販売業者を通して販売可能です。
クラスⅡ以上の医療機器の場合、認証または承認を取得するまで販売はできません。
認証・承認の取得のための審査ではStep3で行った設計開発時の記録や試験・治験等の試験結果が調査されます。
それらにより、当該医療機器の有効性・安全性に問題がないかどうかが確認されます。
また、製造販売業者、製造業者がQMS省令を遵守した適切なQMS体制を整えていることも調査されます。(QMS適合性調査)
当該医療機器の認証・承認後も一貫して品質が良く、有効性・安全性が確保された医療機器が作り続けられる仕組みがあることをここで確認します。